
Eine exergonische Reaktion ist thermodynamisch betrachtet ein Prozess, bei dem die freie Gibbs-Energie abnimmt und dabei nutzbare Energie freigesetzt werden kann. Ich ordne den Begriff hier sauber ein, grenze ihn von exothermen Vorgängen ab und zeige, warum in Chemie und Werkstoffen nicht nur die Energiebilanz, sondern auch Temperatur, Entropie und Reaktionsgeschwindigkeit zählen. Dazu kommen Beispiele aus Batterien, Korrosion und Phasenumwandlungen, weil genau dort die Unterschiede in der Praxis wichtig werden.
Die wichtigsten Punkte auf einen Blick
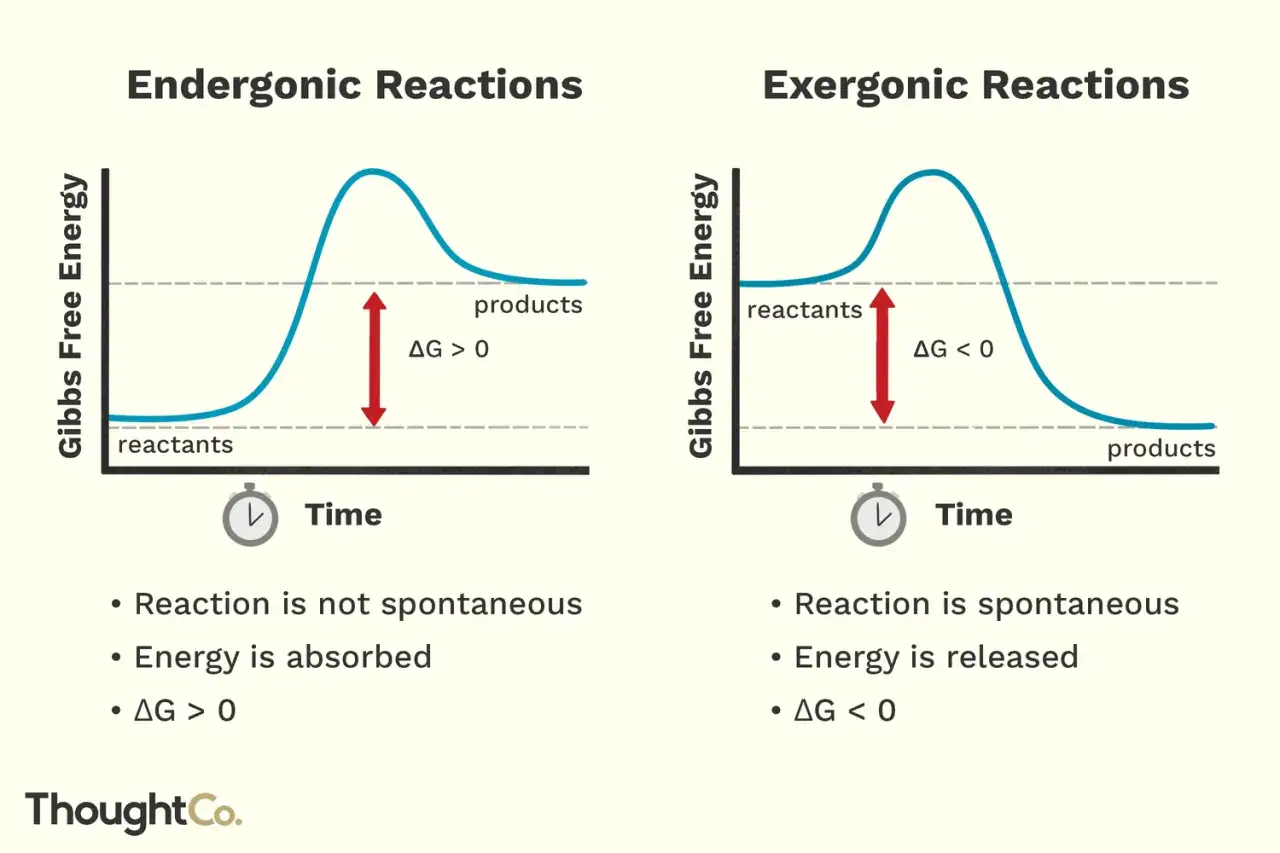
- Eine Reaktion ist thermodynamisch begünstigt, wenn ΔG < 0 gilt.
- Exergonisch bedeutet nicht automatisch exotherm, denn freie Energie und Wärmebilanz sind verschiedene Größen.
- ΔH, ΔS und die Temperatur bestimmen gemeinsam, ob ein Prozess günstig ist.
- Ein negativer ΔG-Wert sagt nichts über die Geschwindigkeit aus.
- In Werkstoffen sind vor allem Elektrochemie, Korrosion, Kristallisation und Phasengleichgewichte relevant.
Was eine exergone Reaktion thermodynamisch ausmacht
Ich trenne diese Begriffe bewusst, weil in der Praxis sonst schnell falsche Erwartungen entstehen. Eine Reaktion ist exergon, wenn die freie Enthalpie des Systems sinkt, also wenn ΔG negativ ist. Das heißt: Unter den gegebenen Bedingungen ist der Ablauf in diese Richtung thermodynamisch begünstigt, bis sich ein Gleichgewicht einstellt.
Wichtig ist der zweite Teil der Aussage: ΔG beschreibt nicht die Geschwindigkeit. Ein Prozess kann energetisch günstig sein und trotzdem sehr langsam verlaufen, wenn die Aktivierungsbarriere hoch ist. Genau deshalb können manche Reaktionen auf dem Papier problemlos ablaufen, im Labor aber ohne Katalysator oder Erwärmung kaum sichtbar sein. Am Gleichgewicht selbst gilt schließlich ΔG = 0; dann gibt es keine treibende Kraft mehr für eine weitere Nettoänderung.
Für die Lesart im Alltag heißt das: Exergonisch beschreibt keine „heftige“ Reaktion, sondern eine Richtung mit energetischem Vorteil. Und genau an dieser Stelle beginnt die häufigste Verwechslung mit exothermen Vorgängen.
Warum exergonisch nicht automatisch exotherm ist
Die Verwechslung ist verständlich, aber fachlich ungenau. Exotherm bezieht sich auf die Enthalpie und damit auf Wärmeabgabe an die Umgebung; exergonisch bezieht sich auf die freie Enthalpie und damit auf die spontane Richtung unter gegebenen Bedingungen. Beides kann zusammenfallen, muss es aber nicht.
| Begriff | Bezug | Typisches Vorzeichen | Aussage | Was oft falsch verstanden wird |
|---|---|---|---|---|
| Exergonisch | Freie Enthalpie | ΔG < 0 | Der Prozess ist thermodynamisch begünstigt. | Wird fälschlich mit schneller Reaktion gleichgesetzt. |
| Endergonisch | Freie Enthalpie | ΔG > 0 | Der Ablauf in diese Richtung braucht Energiezufuhr. | Wird fälschlich als „unmöglich“ missverstanden. |
| Exotherm | Enthalpie | ΔH < 0 | Wärme wird an die Umgebung abgegeben. | Wird fälschlich mit Spontaneität gleichgesetzt. |
| Endotherm | Enthalpie | ΔH > 0 | Wärme wird aus der Umgebung aufgenommen. | Wird fälschlich mit „nicht freiwillig“ gleichgesetzt. |
Der entscheidende Punkt ist der Entropieterm. Wenn der Faktor T·ΔS groß genug ist, kann eine Reaktion trotz positiver Enthalpie dennoch exergonisch sein. Umgekehrt kann ein exothermer Prozess unter ungünstigen Bedingungen endergonisch ausfallen. Ich finde diese Unterscheidung gerade in der Werkstoffkunde wichtig, weil dort Temperaturänderungen nicht nur die Wärmebilanz, sondern auch die Stabilität von Phasen verschieben. Genau das sieht man am besten, wenn man die Gleichung selbst auseinanderzieht.
Wie ΔG aus Enthalpie und Entropie zusammenspielt
Die zentrale Beziehung lautet ΔG = ΔH - TΔS. Sie ist deshalb so nützlich, weil sie zwei unterschiedliche Triebkräfte zusammenfasst: die Enthalpie als Maß für Wärmeeffekte und die Entropie als Maß für die Verteilungsmöglichkeiten eines Systems. Die Temperatur entscheidet dabei, wie stark der Entropieterm gewichtet wird.
Praktisch liest man die Gleichung so: Ein negativer Enthalpiebeitrag hilft der Reaktion, ein positiver Entropiebeitrag hilft ihr ebenfalls, und bei hoher Temperatur gewinnt der Entropieterm an Bedeutung. Deshalb kann ein Prozess bei niedriger Temperatur noch ungünstig sein, bei höherer Temperatur aber plötzlich thermodynamisch günstig werden. Genau aus diesem Grund verändern sich in der Werkstofftechnik auch Stabilitätsbereiche mit der Temperatur so deutlich.
Unter realen Bedingungen kommt noch etwas dazu: Nicht nur Standardgrößen zählen, sondern auch Konzentrationen, Aktivitäten und Druck. Darum ist ΔG° für die Grundidee nützlich, aber im Labor oder in einer technischen Anlage muss ich die tatsächlichen Bedingungen mitdenken. Sobald sich das Reaktionsgemisch verändert, verschiebt sich auch die freie Energie.
Aus der Praxis kenne ich vor allem drei Fragen, die man sich direkt stellen sollte: Ist der Entropiebeitrag groß genug? Ist die Temperatur hoch oder niedrig? Und ist das System überhaupt schon nahe am Gleichgewicht? Wenn diese Punkte klar sind, lassen sich viele scheinbar widersprüchliche Ergebnisse sauber einordnen. Damit landet man schnell bei den Anwendungen, die im Labor und in der Werkstofftechnik wirklich zählen.
Typische Beispiele aus Chemie und Werkstoffen
Am verständlichsten wird der Begriff an konkreten Prozessen. Ich nehme bewusst Beispiele, die in Chemie und Werkstoffkunde wirklich eine Rolle spielen, statt nur Lehrbuchmuster zu wiederholen.
- Batterieentladung - Beim Entladen einer galvanischen Zelle läuft eine redoxchemische Reaktion so ab, dass elektrische Arbeit verfügbar wird. Genau deshalb ist die freie Energieabnahme hier technisch nutzbar.
- Korrosion von Metallen - Rostbildung oder die elektrochemische Auflösung eines Metalls ist thermodynamisch oft begünstigt, auch wenn die Reaktion im Alltag durch Schutzschichten, Sauerstoffmangel oder Diffusionshemmung stark verlangsamt werden kann.
- Kristallisation und Phasenumwandlungen - Wenn sich ein Werkstoff in eine stabilere Kristallstruktur oder eine günstigere Phase umordnet, sinkt oft die freie Energie. Das erklärt, warum Phasengleichgewichte in Legierungen so wichtig sind.
- Oxidbildung bei hohen Temperaturen - In der Hochtemperaturmetallurgie entscheidet die freie Energie der Oxidbildung oft darüber, ob ein Metall stabil bleibt oder bevorzugt mit Sauerstoff reagiert. Für solche Fragen sind freie-Energie-Diagramme besonders hilfreich.
- Verbrennung - Klassisch exotherm und meist auch exergonisch, weil die Produkte energetisch und thermodynamisch tiefer liegen als die Ausgangsstoffe. Der praktische Nutzen liegt hier in der hohen Energieausbeute.
Gerade bei Werkstoffen ist der wichtige Punkt nicht nur, ob ein Prozess möglich ist, sondern wie weit er läuft und welche Mikrostruktur am Ende übrig bleibt. Viele Materialien sind thermodynamisch nicht im absoluten Tiefpunkt, sondern nur metastabil, weil die Umwandlung kinetisch gebremst ist. Genau daraus entstehen oft die Eigenschaften, die wir technisch nutzen wollen.
Woran man in der Praxis erkennt, ob ein Prozess wirklich läuft
Wenn ich eine Reaktion bewerte, schaue ich nie nur auf das Vorzeichen von ΔG. Das wäre zu grob. Entscheidend ist zuerst, ob die Bedingungen stimmen, und danach, ob die Kinetik den Ablauf überhaupt zulässt.
- ΔG unter realen Bedingungen prüfen - Konzentration, Druck, Temperatur und Zusammensetzung können den Wert deutlich verschieben.
- Thermodynamik und Kinetik trennen - Ein negativer ΔG-Wert sagt nur, dass die Richtung günstig ist. Ob die Reaktion schnell genug abläuft, hängt von der Aktivierungsenergie ab.
- Oberflächen und Grenzflächen beachten - In Werkstoffen bestimmen Passivschichten, Korngrenzen und Diffusion oft, ob ein Prozess sichtbar wird oder praktisch blockiert bleibt.
- Katalysatoren richtig einordnen - Sie senken die Aktivierungsbarriere, ändern aber nicht das Gleichgewicht selbst. Das ist ein häufiger Denkfehler.
- Metastabile Zustände mitdenken - Ein Stoff kann in einem Zustand „festhängen“, obwohl ein tieferer energetischer Zustand existiert. Das ist in Gläsern, Legierungen und übersättigten Lösungen normal.
Was in Chemie und Werkstoffen am Ende den Ausschlag gibt
In der technischen Chemie und in der Werkstoffkunde ist die freie Enthalpie kein abstrakter Theoriebegriff, sondern ein Entscheidungswerkzeug. Sie hilft mir zu bewerten, welche Phase stabil ist, welche Reaktionsrichtung bevorzugt wird und warum sich ein Werkstoff unter bestimmten Bedingungen verändert, unter anderen aber stabil bleibt.
Besonders wichtig wird das bei Legierungen, Oxidation, Korrosion und kristallinen Umwandlungen. Auf kleinen Skalen spielen zudem Oberflächen- und Grenzflächenenergien eine größere Rolle, als man zunächst denkt. Deshalb kann ein Material makroskopisch stabil wirken und trotzdem an Defekten, Korngrenzen oder in dünnen Schichten ganz anders reagieren als im Block.
Mein kurzer Merksatz dazu lautet: Wenn du eine exergone Reaktion verstehen willst, lies immer ΔG, ΔH, ΔS und die Temperatur zusammen. Erst dieses Viererbild zeigt, ob ein Prozess thermodynamisch begünstigt ist, wie stark die Triebkraft ausfällt und warum die reale Umsetzung trotzdem Zeit, Katalyse oder besondere Bedingungen braucht.