
Die richtige Methode hängt fast immer von den verfügbaren Daten ab
- Standardbildungsenthalpien liefern meist den saubersten Tabellenwert für Reaktionen unter Standardbedingungen.
- Der Satz von Hess ist ideal, wenn du eine Reaktion aus bekannten Teilreaktionen zusammensetzen musst.
- Kalorimetrie misst den Wärmeeffekt direkt, braucht aber saubere Versuchsbedingungen und Korrekturen.
- Bindungsenthalpien eignen sich für schnelle Überschläge, nicht für präzise Endwerte.
- Vorzeichen, Koeffizienten und Aggregatzustände entscheiden oft darüber, ob das Ergebnis stimmt oder komplett danebenliegt.
- Für Werkstoffe zählt nicht nur der Zahlenwert, sondern auch, wie er sich auf Wärmeabfuhr, Skalierung und Sicherheit auswirkt.
Was die Reaktionsenthalpie in der Praxis bedeutet
Die Reaktionsenthalpie ist die Enthalpieänderung zwischen Edukten und Produkten: ΔH = H(Produkte) - H(Edukte). Unter konstantem Druck entspricht die ausgetauschte Wärme näherungsweise genau diesem Wert, deshalb ist ΔH in der Chemie so viel mehr als nur eine Theoriegröße. Das Vorzeichen ist direkt lesbar: ΔH < 0 bedeutet exotherm, ΔH > 0 bedeutet endotherm.
In der Alltagspraxis trennt man außerdem sauber zwischen Reaktionsenthalpie und Reaktionsenergie. Wenn sich Gase bilden oder verbraucht werden, kann die innere Energie ΔU von ΔH abweichen, weil zusätzlich Volumenarbeit im Spiel ist. Genau deshalb reicht es nicht, einfach „Wärme“ und „Energie“ durcheinanderzuwerfen - die Randbedingungen entscheiden.
Für Standardangaben gilt meist 1 bar und 298 K. Das ist wichtig, weil viele Tabellenwerte nur unter diesen Bedingungen direkt vergleichbar sind. Sobald diese Einordnung steht, stellt sich die eigentliche Frage: Welcher Rechenweg passt zu deinem Datensatz?
Welcher Rechenweg zu welcher Aufgabe passt
Ich trenne die Verfahren in der Praxis zuerst nach der Datenlage, nicht nach der Formel. Das spart Zeit und verhindert, dass man mit einem ungeeigneten Ansatz künstlich Genauigkeit vortäuscht.
| Methode | Worum es geht | Gut geeignet für | Grenze |
|---|---|---|---|
| Standardbildungsenthalpien | Produkte minus Edukte mit tabellierten ΔH°f-Werten | Standardreaktionen, Lehrbuchaufgaben, Stoffdatenrechnung | Nur so gut wie die Tabellenwerte und die Standardbedingungen |
| Satz von Hess | Reaktion aus bekannten Teilreaktionen zusammensetzen | Umwege, fehlende Direktdaten, Umformungen in Aufgaben | Jede Teilgleichung muss korrekt skaliert und gegebenenfalls umgedreht werden |
| Kalorimetrie | Wärme experimentell messen und auf Stoffmenge beziehen | Lösungen, Neutralisationen, Verbrennungen, Laborpraxis | Wärmeverluste, Kalorimeterkonstante und Geräteaufbau beeinflussen das Ergebnis |
| Bindungsenthalpien | Gebrochene Bindungen minus gebildete Bindungen | Schnelle Abschätzung, Plausibilitätscheck | Nur Näherung, vor allem für gasförmige Systeme sinnvoll |
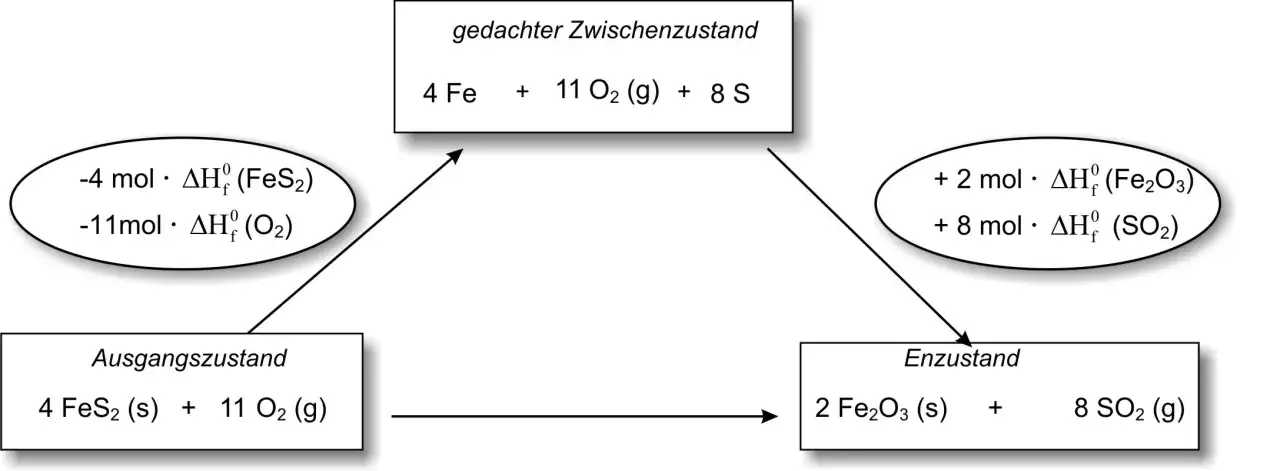
Mit Standardbildungsenthalpien sauber rechnen
Der saubere Tabellenweg ist meistens der direkteste: ΔH°R = Σ ν · ΔH°f(Produkte) - Σ ν · ΔH°f(Edukte). Dabei steht ν für die stöchiometrischen Koeffizienten aus der ausgeglichenen Reaktionsgleichung. Elemente im Standardzustand haben per Konvention eine Bildungsenthalpie von 0.
Lesen Sie auch: Aceton Strukturformel - Was die C=O-Gruppe wirklich bedeutet
Beispiel mit Methanverbrennung
Für die Verbrennung von Methan gilt:
CH4(g) + 2 O2(g) → CO2(g) + 2 H2O(l)
| Stoff | ΔH°f in kJ/mol |
|---|---|
| CH4(g) | -74,8 |
| O2(g) | 0 |
| CO2(g) | -393,5 |
| H2O(l) | -285,8 |
Einsetzen ergibt:
ΔH°R = [ -393,5 + 2 · (-285,8) ] - [ -74,8 + 2 · 0 ] = -890,3 kJ/mol
Das ist die Reaktionsenthalpie pro mol Methan für die Gleichung, wie sie oben geschrieben steht. Genau an dieser Stelle machen viele den ersten typischen Fehler: Sie vergessen, dass der Zahlenwert an die stöchiometrische Schreibweise gebunden ist.
Noch wichtiger für die Praxis ist der Aggregatzustand. Wenn bei derselben Verbrennung Wasser als Dampf statt als Flüssigkeit entsteht, ändert sich der Wert deutlich. Das ist kein Detail, sondern ein Hinweis darauf, dass der thermische Effekt des Systems stark von den Randbedingungen abhängt.
Wenn du keine passenden Tabellenwerte für die Reaktion selbst findest, ist der nächste Weg der Satz von Hess. Genau dafür ist er gedacht.
Der Satz von Hess macht Zwischenstufen egal
Der Satz von Hess beruht darauf, dass die Enthalpie eine Zustandsgröße ist. Für die Rechnung heißt das: Es ist egal, auf welchem Weg Produkte aus Edukten entstehen, solange Start- und Endzustand gleich bleiben. Genau das macht ihn so nützlich, wenn eine Reaktion nicht direkt gemessen oder tabelliert ist.
Die drei Rechenregeln sind schlicht:
- Wenn du eine Gleichung umdrehst, wechselt das Vorzeichen von ΔH.
- Wenn du eine Gleichung mit einem Faktor multiplizierst, multiplizierst du ΔH mit demselben Faktor.
- Wenn du Teilgleichungen addierst, addierst du auch ihre Enthalpien.
Ein kurzes Beispiel ist die Bildung von Kohlenmonoxid:
C(graphit) + 1/2 O2(g) → CO(g)
Direkt braucht man dafür nicht immer einen Tabellenwert, aber man kann zwei bekannte Reaktionen nutzen:
C(graphit) + O2(g) → CO2(g) ΔH = -393,5 kJ/mol
CO(g) + 1/2 O2(g) → CO2(g) ΔH = -283,0 kJ/mol
Die zweite Gleichung drehe ich um und addiere sie zur ersten. Dann ergibt sich:
C(graphit) + 1/2 O2(g) → CO(g) ΔH = -110,5 kJ/mol
Genau dieser Mechanismus ist in vielen Aufgaben entscheidend: Nicht der direkte Weg zählt, sondern die saubere Bilanz. Wenn du experimentell arbeitest oder echte Stoffmengen misst, kommst du aber schnell zu einer anderen Frage: Was ist der Wärmeeffekt im realen Aufbau?
Kalorimetrie zeigt den realen Wärmeeffekt
Bei der Kalorimetrie messe ich nicht den Tabellenwert, sondern die tatsächlich frei werdende oder aufgenommene Wärme. Für Lösungen und viele Schul- oder Laborversuche gilt näherungsweise q = m · c · ΔT, also Masse mal spezifische Wärmekapazität mal Temperaturänderung. Die Reaktionsenthalpie ergibt sich dann aus dem negativen des gemessenen Wärmebetrags, bezogen auf die umgesetzte Stoffmenge.
Ein einfaches Beispiel: 100,0 g Lösung erwärmen sich um 3,0 K, die Wärmekapazität ist wie bei Wasser ungefähr 4,18 J/(g·K), und umgesetzt wurden 0,0200 mol. Dann nimmt die Lösung etwa 1,254 kJ auf, also liegt die Reaktion näherungsweise bei ΔH ≈ -62,7 kJ/mol. In echten Messungen addiere ich die Kalorimeterkonstante separat dazu, wenn sie nicht vernachlässigbar ist.
Wichtig ist auch der Gerätetyp. In einem einfachen Becherkalorimeter arbeite ich unter konstantem Druck, also komme ich relativ direkt zu ΔH. Im Bombenkalorimeter messe ich dagegen unter konstantem Volumen zunächst ΔU. Wenn Gase beteiligt sind, braucht man dann eine Umrechnung, oft näherungsweise über ΔH = ΔU + ΔngasRT.
Kalorimetrie ist deshalb stark, weil sie reale Systeme abbildet. Gleichzeitig ist sie empfindlich gegenüber Wärmeverlusten, Rührfehlern und schlecht bestimmter Kalorimeterkonstante. Für schnelle Abschätzungen ist deshalb noch ein anderer Weg nützlich: die Bindungsenthalpien.
Bindungsenthalpien sind gut für Überschläge
Wenn mir nur eine grobe Abschätzung reicht, rechne ich oft mit mittleren Bindungsenthalpien. Das Prinzip ist einfach: ΔH ≈ Summe der gebrochenen Bindungen - Summe der gebildeten Bindungen. Je mehr Energie ich für das Aufbrechen der Ausgangsbindungen brauche und je mehr Energie bei der Bindungsbildung frei wird, desto stärker fällt die Reaktion exo- oder endotherm aus.
Ein klassisches Beispiel ist:
H2(g) + Cl2(g) → 2 HCl(g)
Mit typischen Mittelwerten von etwa 436 kJ/mol für H-H, 243 kJ/mol für Cl-Cl und 431 kJ/mol für H-Cl ergibt sich:
436 + 243 - 2 · 431 = -183 kJ/mol
Das liegt nah an der Realität, ist aber eben ein Überschlag. Genau deshalb nutze ich Bindungsenthalpien für Plausibilitätsprüfungen, nicht als endgültigen Designwert. Bei Flüssigkeiten, Feststoffen, Resonanzsystemen oder stark wechselwirkenden Umgebungen werden die Abweichungen schnell größer.
Damit landet man fast automatisch bei der Frage, welche Fehler die Rechnung am häufigsten kippen. Dort lohnt sich ein sehr nüchterner Blick.
Diese Fehler kosten fast immer Genauigkeit
Die meisten falschen Ergebnisse entstehen nicht durch komplizierte Chemie, sondern durch einfache Ungenauigkeit im Ansatz. Ich prüfe deshalb immer dieselben Punkte:
| Fehler | Folge | So vermeidest du ihn |
|---|---|---|
| Vorzeichen vertauscht | Exotherm und endotherm werden verwechselt | Merke dir: Wärmeabgabe ist negativ, Wärmeaufnahme positiv |
| Koeffizienten nicht mitgerechnet | Der molare Wert stimmt nicht | Jede Zahl vor der Formel in die Rechnung übernehmen |
| Aggregatzustand ignoriert | Deutliche Abweichung vom Tabellenwert | Immer s, l, g oder aq mitlesen |
| kJ und kJ/mol vermischt | Falsche Skalierung der Aussage | Sauber notieren, worauf sich der Wert bezieht |
| Kalorimeterwärme weggelassen | Messwert wird systematisch zu klein | Kalorimeterkonstante und Wärmeverluste berücksichtigen |
| ΔU mit ΔH gleichgesetzt | Verbrennungs- und Gasreaktionen werden falsch bewertet | Bei konstantem Volumen die Umrechnung prüfen |
Der Aggregatzustand ist kein Detail, sondern oft der Punkt, an dem die ganze Bilanz kippt. Das ist besonders wichtig, wenn man Werte aus Tabellen in reale Prozesse überträgt. Genau dort zeigt sich, warum die Reaktionsenthalpie für Werkstoffe und Prozessführung mehr ist als eine Schulzahl.
Warum der Wert für Werkstoffe und Prozessführung so wichtig ist
In der Werkstoffchemie schaue ich auf die Reaktionsenthalpie nie isoliert. Eine stark exotherme Reaktion kann im Labor noch bequem aussehen, im größeren Ansatz aber zu lokalen Überhitzungen, ungleichmäßiger Aushärtung oder Sicherheitsproblemen führen. Umgekehrt bedeuten endotherme Prozesse oft höhere Energiezufuhr, längere Heizzeiten oder engere Temperaturführung.
- Bei Harzen und Klebstoffen ist die Wärmefreisetzung für Aushärtung und Bauteilverzug relevant.
- Bei Oxidations- oder Verbrennungsprozessen entscheidet ΔH mit darüber, wie viel Kühlung nötig ist.
- Bei Kalkination, Zersetzung oder bestimmten Synthesen muss die Wärme überhaupt erst bereitgestellt werden.
- Bei der Skalierung von Labor auf Anlage prüfe ich nicht nur den Zahlenwert, sondern auch Wärmeabfuhr, Rührleistung und Temperaturspitzen.
Die wichtigste Einordnung bleibt aber: Eine negative Reaktionsenthalpie sagt nichts über die Geschwindigkeit aus. Eine Reaktion kann stark exotherm und trotzdem langsam sein, wenn die Aktivierungsenergie hoch ist. Wer die Zahl also wirklich nutzen will, liest sie immer zusammen mit Kinetik, Stoffdaten und Prozessrandbedingungen. Genau das macht aus einer reinen Formel eine brauchbare Entscheidungshilfe.